
Preparing for an FDA Inspection: A Comprehensive Guide for 503B Outsourcing Facilities
503B outsourcing facilities compound sterile drugs and other products for use in clinics, hospitals and health systems. The FDA has implemented strict oversight procedures that include regular inspections. The guidance document emphasizes that the FDA’s approach is risk-based, meaning that the scope and intensity of an inspection will depend on a facility’s compliance history, the complexity of its operations, and its overall risk profile.
An FDA inspection is not merely a checklist exercise; it is an opportunity for facilities to demonstrate a robust commitment to patient safety and product quality. Preparing for an inspection requires a thorough review of all systems and processes, as well as a proactive approach to identifying and addressing potential gaps before an inspector arrives.
1. Quality Systems and Management Oversight
A cornerstone of compliance for any 503B facility is a robust quality system that encompasses management oversight, risk assessment, and continuous improvement. Here are the key points to consider:
Leadership and Governance: Senior management must demonstrate visible commitment to quality. This includes establishing clear roles and responsibilities, holding regular quality review meetings, and ensuring that quality is prioritized across all levels of the organization.
Quality Metrics and Risk Management: Facilities should implement quality metrics that allow for the early detection of trends or deviations. A risk-based approach to both production and process controls ensures that issues are identified and addressed proactively.
Corrective and Preventive Actions (CAPA): A well-documented CAPA system is critical. It must not only address identified issues but also track the effectiveness of corrective actions over time.
Internal Audits and Self-Inspections: Regular internal audits help assess compliance with written procedures and regulatory requirements. Documentation of these audits, along with follow-up actions, should be easily accessible for inspectors.
A well-functioning quality system reassures inspectors that the facility is continually monitoring its processes and is prepared to address any deviations immediately.
2. Facilities, Equipment, and Environmental Controls
The physical environment where compounding takes place is central to ensuring product safety and quality. FDA inspectors look closely at the layout, maintenance, and operation of the facility.
Facility Design and Layout: The design of the facility should support unidirectional flow to prevent cross-contamination. Critical areas—such as cleanrooms and sterile compounding zones—must meet strict environmental standards.
Equipment Qualification and Calibration: All equipment should be properly qualified (i.e., Installation, Operational, and Performance Qualification) and routinely calibrated. Preventive maintenance schedules must be documented and adhered to.
Environmental Monitoring: Regular environmental monitoring in critical areas is essential. This includes routine sampling of air, surfaces, and water used in the compounding process. The facility must have documented action limits and procedures for addressing excursions.
Contamination Controls: Inspections will assess the physical controls in place to prevent contamination—from HVAC systems to controlled entry protocols. The facility should also demonstrate that it is prepared for unplanned events (such as HVAC failures) through robust contingency planning.
By ensuring that facilities and equipment are managed according to these principles, an outsourcing facility can significantly reduce the risk of non-compliance and product contamination.
3. Personnel Training and Qualifications
The FDA places a high priority on the competence and training of personnel involved in compounding and quality control. This system is a critical component of the overall quality infrastructure.
Training Programs: All personnel must receive initial and ongoing training that covers both the theoretical and practical aspects of their roles. Training should include aseptic techniques, hygiene, proper gowning, and emergency response.
Competency Assessments: Periodic evaluations of personnel performance ensure that skills remain current. Records of these assessments, along with refresher training sessions, should be maintained.
Clear Role Definition: Every employee’s responsibilities should be clearly documented. This includes job descriptions, training records, and qualification documents.
Cross-Training: In many facilities, redundancy is crucial. Cross-training employees on multiple tasks can minimize disruptions during inspections or when key personnel are absent.
Hygiene and Health: Given the sterile nature of many compounded products, personal hygiene protocols are non-negotiable. Regular health assessments and adherence to policies regarding illness are essential.
Proper training and clear documentation not only ensure that staff members are capable of maintaining compliance but also demonstrate to inspectors that the facility values and invests in its human capital.
4. Production and Process Controls
The production process is at the heart of a 503B facility’s operations. FDA guidance stresses that robust process controls are essential for ensuring that products are manufactured consistently and safely.
Standard Operating Procedures (SOPs): Every step in the production process should be governed by written SOPs. These must be clear, current, and reflect actual practices.
Process Validation: Critical manufacturing processes should be validated to ensure that they consistently produce a product meeting its predetermined specifications. Validation data must be documented comprehensively.
In-Process Controls: Routine monitoring and controls should be integrated into production operations. This includes verifying critical parameters (such as temperature, pressure, and mixing times) at various stages of production.
Batch Record Review: Complete and accurate batch records are essential. They provide a traceable history for every batch produced and allow inspectors to verify that production controls were in place.
Change Control Procedures: Any change in process, equipment, or raw materials must be managed through a formal change control system. The impact of changes must be assessed, documented, and approved by quality management before implementation.
By focusing on production and process controls, facilities can ensure that every product is manufactured to the highest quality standards and that any deviations are promptly identified and rectified.
5. Laboratory Controls and Testing
The laboratory functions within a 503B facility are critical to verifying that products meet quality specifications. FDA guidance emphasizes the importance of robust laboratory controls.
Method Validation: All analytical methods used for product testing must be validated. This includes demonstrating that methods are specific, accurate, and reproducible.
Routine Testing and In-Process Checks: Regular testing of raw materials, in-process samples, and finished products is required. Laboratories must have well-documented procedures for sampling, testing, and reporting.
Out-of-Specification (OOS) Procedures: If test results fall outside of predefined specifications, the facility must have a robust investigation process. Documentation should detail the steps taken to resolve the issue.
Equipment and Reagent Controls: The testing equipment must be routinely calibrated and maintained, and reagents must be tracked to ensure their validity and reliability.
Quality Control Samples: The use of control samples and proficiency testing helps ensure that the laboratory is operating within defined parameters. These records provide additional assurance that the laboratory’s processes are reliable.
A strong laboratory control system not only ensures product quality but also serves as a critical line of defense in identifying potential issues before products reach patients.
6. Documentation and Record Keeping
Accurate, complete, and timely documentation is the lifeblood of regulatory compliance. The FDA’s inspection guidance makes it clear that every process, decision, and corrective action must be documented.
Comprehensive SOPs and Protocols: Written procedures must be available for every aspect of the facility’s operations—from quality management to production and laboratory testing.
Batch Records and Logbooks: Complete records of every batch produced, including any deviations and corrective actions taken, must be maintained and easily accessible during an inspection.
Electronic Records and Data Integrity: For facilities using electronic systems, data integrity is paramount. This includes implementing access controls, audit trails, and secure data storage solutions.
Training Records: Documentation of all training sessions, competency evaluations, and qualifications for personnel should be maintained.
CAPA Documentation: Records of investigations, root cause analyses, and CAPA initiatives should be thorough and reflect a commitment to continuous improvement.
Retention Policies: Clear record retention policies ensure that all documents are kept for the required duration and can be retrieved quickly during an inspection.
Effective documentation not only supports day-to-day operations but also provides a clear, audit-ready trail that demonstrates compliance with regulatory requirements.
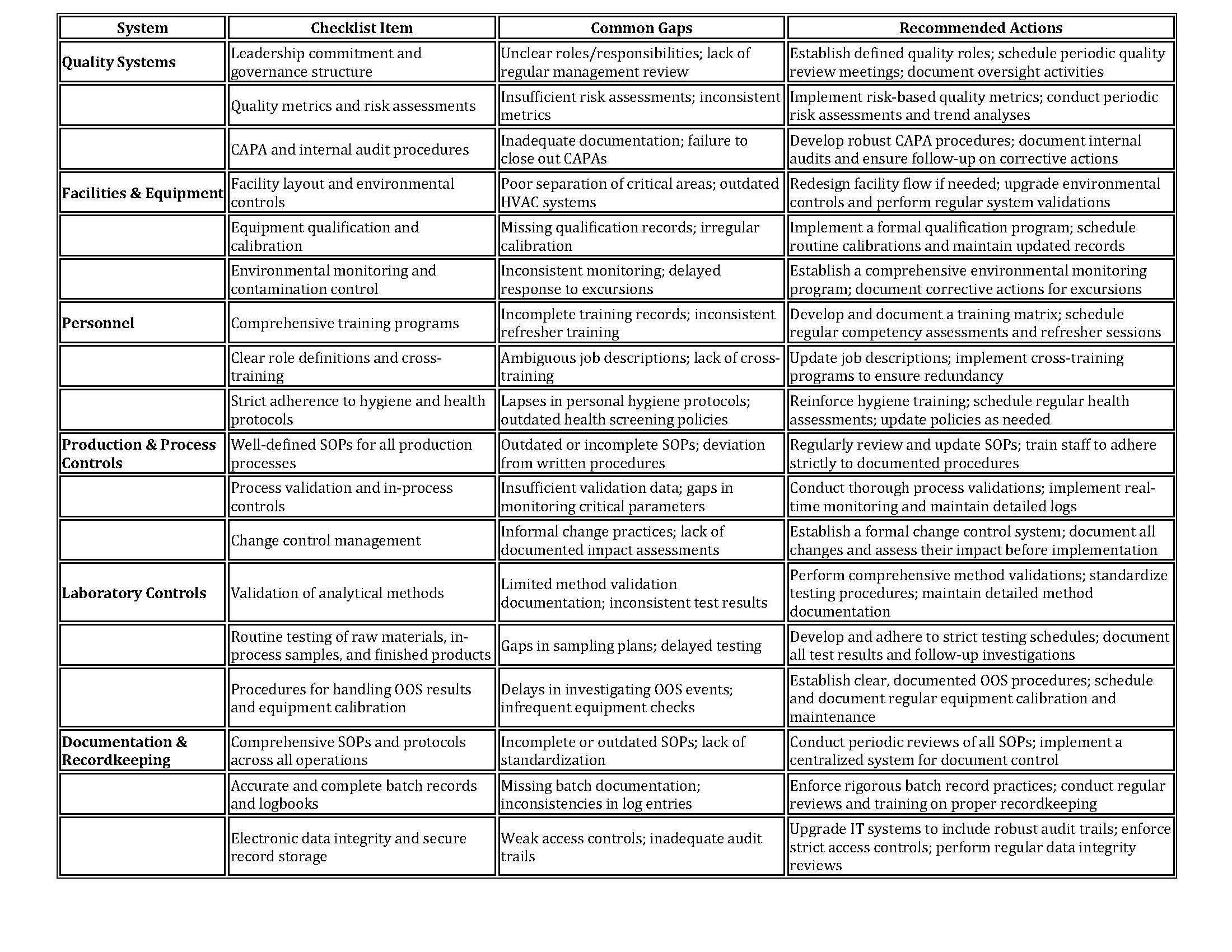
Gap Analysis Checklist Table Organized by Six Systems
The table below provides a practical gap analysis checklist that can help facilities identify potential areas for improvement. For each of the six systems, key requirements are listed along with common gaps and recommended actions.
Conclusion
Preparing for an FDA inspection as a 503B outsourcing facility involves far more than tidying up documents on the shelf. It requires a strategic, facility-wide commitment to quality, safety, and regulatory excellence. By focusing on the six critical systems—Quality Systems, Facilities & Equipment, Personnel, Production & Process Controls, Laboratory Controls, and Documentation & Recordkeeping—facilities can not only meet FDA expectations but also ensure that they deliver safe, high-quality products to patients.
A proactive approach to self-inspection and gap analysis is key. The checklist provided above serves as a practical tool for identifying potential vulnerabilities and implementing corrective measures well before an inspection occurs. Ultimately, the goal is to create a culture of continuous improvement where every process is regularly evaluated, risks are mitigated, and the entire organization is aligned in the mission to uphold the highest standards in pharmaceutical compounding.
By embracing these strategies, 503B outsourcing facilities can be confident in their readiness for FDA inspections—and more importantly, in their ability to consistently deliver safe, effective, and high-quality compounded products.
Looking for an FDA Readiness Partner?
If you’re ready to bring quality to the center of your 503B or advanced therapy operations, consultants at Restore Health Consulting LLC can help with:
Equipment IQ/OQ/PQ Protocols, Execution, & Reports
Utility Systems
Cleanroom EMPQ (Environmental Baseline Study)
Cleaning Validation
Disinfectant Efficacy Study
21 CFR Part 11 Computer System Validation (CSV)
Audits & Gap Assessments
GMP Training
And More
Disclaimer:
The information provided on this website does not, and is not intended to, constitute legal advice; instead, all information, content, and materials available on this site are for general informational purposes only. Information on this website may not constitute the most up-to-date legal or other information. Readers of this website should contact their attorney to obtain advice with respect to any particular legal matter.